ВВЕДЕНИЕ
Квантовая механика и статистическая физика в принципе позволяют дать исчерпывающее объяснение любым экспериментальным данным о реакционной способности органических соединений и предсказать возможные направления реакций. Однако для реализации этих возможностей необходимо располагать очень мощными ЭВМ и достаточно точными вычислительными методами. За последние годы в этих областях был достигнут существенный прогресс. Химические институты получили возможность широко использовать быстродействующие ЭВМ, а благодаря быстрому развитию квантовой химии были разработаны достаточно эффективные полуэмпирические и неэмпирические варианты метода молекулярных орбиталей (МО), которые можно использовать для изучения реакционной способности больших молекул, представляющих интерес не только для органической химии, но даже для биохимии. С их помощью удается установить, какие факторы определяют направление и относительный выход продуктов реакции, а также получить недоступную для эксперимента информацию о геометрии и электронной структуре переходных состояний. Однако в условиях многообразия и неравноценности квантовохимических методов представляется целесообразным охарактеризовать конкретные возможности каждого из них, отметить преимущества и недостатки и оценить перспективы развития путей их применения в органической химии.
Подробное описание теории МО можно найти в целом ряде монографий (см., например, [1, 2]). Однако при проведении прикладных квантовохимических расчетов нет необходимости углубляться в изучении ее основ. Необходимо иметь лишь общее представление о теории МО, уметь пользоваться квантовохимическими программами и научиться извлекать полезную информацию из результатов расчета. Поэтому в настоящем разделе дано лишь краткое описание теории МО и расшифрованы сокращенные названия методов, которые наиболее широко используются в отечественной и зарубежной литературе в области органической химии.
В основе современной квантовой химии лежит уравнение Шрёдингера для стационарных состояний. Его обычно решают в адиабатическом приближении, т.е. в предположении, что ядерную и электронную волновые функции можно разделить и решать уравнения для движения ядер и электронов раздельно. В этом приближении уравнение Шрёдингера для электронной волновой функции записывается следующим образом:
ĤΨ=ЕΨ, (1)
где Ĥ — гамильтониан системы, т.е. сумма операторов кинетической и потенциальной энергий; Ψ=Ψ(х1, x2, …, хn) — волновая функция для системы из n частиц, которая зависит от их расположения в пространстве и спинов; Е — полная электронная энергия. Однако точно решить это уравнение удается лишь в случае одноэлектронных систем. Поэтому в квантовохимических расчетах используются приближенные методы. Среди них наиболее широкое распространение получил метод Хартри—Фока [1, 2], или метод самосогласованного поля (ССП). В этом методе полагается, что каждый электрон движется в поле атомных ядер, положение которых фиксировано в пространстве, и в эффективном (усредненном) поле других электронов. Многоэлектронную волновую функцию ищут в виде антисимметризованнрго произведения спин-орбиталей, т.е. одноэлектронных МО φi(μ), умноженных на спиновые волновые функции α или β соответствующего электрона:
Ψ=Adet[φ1(1)α(1)φ1(1')β(1')…φn(n')β(n')], (2)
где 2n — число электронов в молекуле; μ — номер электрона. Значение коэффициента А определяется условием нормировки |Ψ|2 к единице. В приближении Хартри—Фока уравнение Шрёдингера переходит в систему интегрально-дифференциальных уравнений для движения каждого отдельного электрона:
Fφi=εiφi, (3)
где F — фокиан (гамильтониан в приближении ССП), εi — энергии МО.
Молекулярные орбитали φi(μ) обычно ищут в виде линейных комбинаций атомных орбиталей (ЛКАО) χi(μ)
φm(μ)=∑Cmiχi(μ), (4)
где Cmi — искомые коэффициенты.
Таким образом, совокупность атомных орбиталей (АО) χiявляется физически наглядным базисом для построения молекулярной волновой функции φm. Обычно количество базисных АО недостаточно велико, т.е. базис является неполным. Основное требование к неполному базисному набору АО заключается в том, чтобы он мог достаточно точно передать распределение электронной плотности в молекуле, т.е. базис должен быть достаточно сбалансированным. К сожалению, на практике выполнить это условие бывает трудно. Атомные орбитали хорошо аппроксимируют распределение электронной плотности в изолированных атомах, но их использование при расчетах волновых функций молекул может приводить к погрешностям, связанным с неспособностью передать некоторые особенности изменения электронного распределения при образовании валентных связей. Поэтому в случае молекул приходится расширять атомные базисные наборы χiпутем включения в них дополнительных функций.
Для определения коэффициентов Cmiиспользуется вариационная процедура минимизации полной электронной энергии молекулы Е. Рутан показал [З], что коэффициенты Cmi, соответствующие минимальной величине, могут быть определены из следующей системы уравнений:
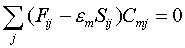 , (5)
, (5)
где
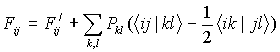 ; условие нормировки
; условие нормировки 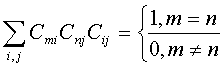
Суммирование ведется по всем базисным орбиталям χi, χj, χkи χl;
Sij — интервал перекрывания АО χi и χj, F'ij — матричный элемент одноэлектронного гамильтониана, в который включены кинетическая энергия электронов и энергия взаимодействия электронов и атомных ядер; Pkl — матрица зарядов и порядков связей; <ij|kl> — интеграл кулоновского взаимодействия двух электронов:
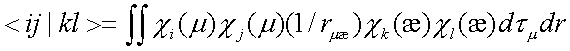 . (6)
. (6)
В этом выражении интегрирование производится по всему пространству декартовых координат: rμæ — расстояние между электронами μ и æ.
Для решения нелинейной системы уравнений (5) применяется метод самосогласования, в котором в качестве нулевого приближения берется совокупность коэффициентов Cmi, причем выбор их может быть довольно произвольным. По коэффициентам нулевого приближения строят матрицу Fij; по ней, решая уравнения (5), находят набор коэффициентов Cmi, который используют для построения новой матрицы Fijи т.д., пока матричные элементы Fijи коэффициенты Cmi не перестанут изменяться.
На практике обычно пользуются как полуэмпирическими, так и неэмпирическими методами. Они различаются методикой вычисления матричных элементов, описывающих взаимодействие электронов между собой и электронов и атомных ядер в уравнениях (5). В полуэмпирических методах для этой цели используются приближенные эмпирические формулы и известные из эксперимента параметры атомов. В неэмпирических методах проводится непосредственный аналитический расчет матричных элементов. Для неспециалиста название "неэмпирический" является синонимом слова "точный", но в действительности это не так. Неэмпирические методы также являются приближенными, прежде всего из-за неполноты использованного базиса. Большинство неэмпирических расчетов проводят в базисах небольшого и среднего размеров. Это вносит существенную ошибку в результаты расчета, так как такие базисные наборы не могут передать некоторые особенности распределения электронной плотности в молекулах. В результате все без исключения параметры молекул вычисляются с некоторой ошибкой. Было бы желательно (хотя на практике это сделать очень трудно из-за больших затрат машинного времени) использовать достаточно большие базисы, которые обеспечивают выход на так называемый хартри-фоковский предел, когда дальнейшее увеличение числа базисных орбиталей не влияет на полученные результаты. Но даже в этом случае мы найдем не точное решение уравнения Шрёдингера, а лишь его решение в приближении Хартри-Фока. Для получения точного решения уравнения Шрёдингера необходимо еще учесть электронную корреляцию (см. ниже).
Полуэмпирические расчеты в настоящее время чаще всего проводят в валентных приближениях ППДП, ЧПДП и ПДДП [4].
В перечисленных выше приближениях волновые функции рассчитывают только для валентных электронов, а электроны внутренних оболочек включают в остов молекулы; используют минимальный слейтеровский базис; пренебрегают некоторой частью матричных элементов кулоновского взаимодействия электронов. Последнее допущение является наиболее существенным, так как оно позволяет значительно упростить расчет. Количество матричных элементов кулоновского взаимодействия электронов (6) очень велико, оно пропорционально числу базисных орбиталей в четвертой степени, поэтому при увеличении базиса в 2 раза необходимо вычислить в 16 раз больше кулоновских интегралов (6). Их расчет требует очень больших затрат машинного времени и для многих соединений, представляющих интерес с точки зрения органической химии, на современных ЭВМ практически неосуществим.
В полуэмпирических методах пренебрегают большей частью кулоновских интегралов (6), которые имеют небольшую абсолютную величину, но точность расчета при этом заметно снижается (величина каждого кулоновского интеграла, которым пренебрегают, мала, но их количество велико). Это удается частично компенсировать за счет удачного подбора параметров. В приближении ППДП учитываются только одноцентровые интегралы типа <ii|ii> и двухцентровые интегралы типа <ii|kk> (χi и χk принадлежат разным атомам). В приближении ЧПДП дополнительно учитываются кулоновские интегралы, у которых все четыре орбитали χi, χj, χkи χlпринадлежат одному атому. В приближении ПДДП, кроме интегралов, которые учитываются в приближениях ППДП и ЧПДП, в расчет дополнительно включаются интегралы <ik|jl>, у которых орбитали χi и χkпринадлежат одному атому, а χj и χl— другому.
Для вычисления одноцентровых матричных элементов в полуэмпирических методах, как правило, используются данные эксперимента по атомным спектрам, а двухцентровые обычно рассчитывают по эмпирическим формулам, которые приближенно отражают зависимость взаимодействия электронов и атомных ядер и электронов между собой от взаимного расположения атомных орбиталей.
Для определения параметров двухцентровых интегралов, входящих в эмпирические формулы, приходится привлекать данные не только для атомов, но и для малых молекул. При этом задача решается неоднозначно и различия в способах подбора параметров характеризуют модификации методов ППДП, ЧПДП и ПДДП. Рациональней подбор параметров позволяет смягчить недостатки приближений ППДП, ЧПДП и ПДДП и даже самого метода ССП.
Для расчета свойств основного состояния органических молекул в настоящее время широко используются следующие полуэмпирические методы: ППДП/2 [5, 6] и ППДП/БУ [7] (приближение ППДП), ЧПДП [8] и различные варианты метода МЧПДП [9 — 13] (приближение ЧПДП), МПДП и AM1 [14—16] (приближение ПДДП). Параметры в методах ППДП/2 и ЧПДП подобраны так, чтобы воспроизвести распределение электронной плотности в малых молекулах, которое дает неэмпирический расчет в минимальном слейтеровском базисе. Эти методы позволяют рассчитать геометрию и дипольные моменты молекул. Их можно использовать для расчета индексов реакционной способности, но для вычисления поверхностей потенциальной энергии органических реакций и параметров переходных состояний данные методы не годятся.
Параметры в методе ППДП/БУ подобраны так, чтобы воспроизвести кривые Морзе для двухатомных молекул. Этот метод позволяет рассчитать геометрию, потенциалы ионизации и энергии атомизации молекул, но хорошее согласие с экспериментом получается только для малых молекул. С увеличением размера молекулы расхождение данных расчета и эксперимента становится больше. Кроме того, метод ППДП/БУ существенно переоценивает стабильность переходных состояний для большинства органических реакций. Параметры, которые используются в методах МЧПДП/2, МЧПДП/3, МПДП и AM1 для оценки величины матричных элементов, подобраны так, чтобы воспроизвести геометрию и теплоты образования органических соединений. Кроме геометрии и теплот образования, эти методы позволяют неплохо вычислить потенциалы ионизации и дипольные моменты молекул, а также геометрию переходных состояний и энергии активации. В настоящее время их довольно широко используют для расчета поверхностей потенциальной энергии органических реакций. В большинстве случаев получаются удовлетворительные результаты. При вычислении геометрии и относительной стабильности интермедиатов и переходных состояний метод МПДП обычно дает лучшее согласие с экспериментом по сравнению с методами МЧПДП/2 и МЧПДП/3. Это связано, по-видимому, с более точным учетом кулоновского взаимодействия электронов.
Расчетов с использованием метода AM1 пока опубликовано очень мало, поэтому мы не можем охарактеризовать область его применения. По-видимому, он дает примерно такие же результаты, как метод МПДП. Метод AM1 отличается от МПДП способами подбора параметров и формулой для вычисления энергии отталкивания остова. За счет более совершенной методики подбора параметров удается немного повысить точность вычисления теплот образования молекул, содержащих атомы кислорода и азота. За счет изменения формулы для вычисления энергии отталкивания остова удалось несколько улучшить согласие с экспериментом при расчете межмолекулярных взаимодействий. Однако количественное улучшение согласия с экспериментом, которое при этом получается, по имеющимся в настоящее время данным невелико.
В неэмпирических методах все матричные элементы взаимодействия электронов и атомных ядер и электронов между собой вычисляются с помощью аналитического расчета необходимых интегралов в некотором базисе АО. Наиболее точно распределение электронной плотности в атомах можно передать с помощью слейтеровских АО, т.е. функций типа ехр(-αr), rехр(-αr), хехр(-αr), yехр(-αr) и т.д. Однако со слейтеровскими орбиталями очень трудно вычислить интегралы, которые входят в фокиан для молекул. Поэтому в качестве базисных АО обычно берут гауссовы функции — для s-орбиталей: ехр(-αr2);
для p-орбиталей: xехр(-αr2), yехр(-αr2), zехр(-αr2);
для d-орбиталей: x2exp(-αr2), y2exp(-αr2), z2exp(-αr2), xyexp(-αr2), xzexp(-αr2), yzexp(-αr2).
Это так называемые примитивные гауссовые функции. С ними относительно легко вычислять матричные элементы, но, когда их мало, они плохо воспроизводят распределение электронной плотности в атомах и молекулах. В связи с этим гауссовых орбиталей приходится брать намного больше, чем слейтеровских. Обычно используют так называемые сгруппированные базисы, в которых каждая базисная орбиталь представляет собой линейную комбинацию из нескольких примитивных гауссовых функций.
В настоящее время для изучения реакционной способности и строения органических соединений наиболее широко используются базисы, предложенные Поплом с сотр.: минимальный базис ОСТ-3ГФ, валентно-расщепленные базисы 3-21ГФ, 4-31ГФ, 6-31ГФ, валентно-расщепленные базисы с поляризационными орбиталями 6-31ГФ* и 6-31ГФ**, валентно-расщепленные базисы с диффузными s- и p-орбиталями 3-21+ГФ и 4-31+ГФ [17—21].
ОСТ-ЗГФ — минимальный базис, в котором каждая слейтеровская орбиталь аппроксимирована линейной комбинацией из трех гауссовых функций.
3-21ГФ, 4-31ГФ, 6-31ГФ — валентно-расщепленные базисы, в которых каждая слейтеровская орбиталь для электронов внутренних оболочек аппроксимирована линейной комбинацией соответственно из трех, четырех и шести гауссовых функций, а для валентных электронов вместо каждой слейтеровской орбитали используются две линейные комбинации гауссовых функций: одна — из двух (базис 3-21ГФ) или трех (базисы 4-31ГФ и 6-31ГФ), а другая — из одной примитивной гауссовой функции. В этих трех базисах одинаковое число сгруппированных гауссовых функций, но число примитивных функций в базисах 6-31ГФ и 4-31ГФ больше, чем в базисе 3-21ГФ.
Использование базисов 6-31ГФ и 4-31ГФ позволяет получить более точную аппроксимацию распределения электронной плотности, особенно вблизи атомных ядер, однако точность результатов при вычислении большинства физико-химических параметров молекул повышается незначительно, а затраты машинного времени возрастают существенно (наиболее сильно в расчетах с оптимизацией геометрии). Поэтому в прикладных работах в настоящее время наиболее широко используется базис 3-21ГФ.
6-31ГФ* и 6-31ГФ** - валентно-расщепленные базисы с поляризационными d-орбиталями. В базис 6-31ГФ* включены поляризационные d-орбитали на р-элементах, т.е. на атомах углерода, азота и т.д., а в базис 6-31ГФ** — d-орбитали на р-элементах и р-орбитали на атомах водорода. Включение в базис поляризационных орбиталей существенно повышает точность расчета, но, к сожалению, при этом очень сильно возрастают затраты машинного времени.
3-21+ГФ и 4-31+ГФ – с дополнительными диффузными s- и р-орбиталями – используются в квантовохимических расчетах физико-химических параметров анионов. При образовании аниона из электрически нейтральной молекулы МО становятся более диффузными. Этот эффект не удается передать при использовании валентно-расщепленных базисов, подобранных для расчета незаряжанных молекул, поэтому точность неэмпирического расчета для анионов в таких базисах оказывается существенно ниже, чем для нейтральных соединений. Повышение точности может быть достигнуто за счет дополнительного включения в базис диффузных s- иp-орбиталей. Это наиболее экономичный способ повышения точности неэмпирических расчетов для анионов. Затраты машинного времени при этом возрастают лишь в 1,5 раза. При изучении анионов можно также использовать базисы с поляризационными орбиталями, так как d-орбитали на р-элементах и р-орбитали на атомах водорода являются диффузными функциями, но в этом случае затраты машинного времени на выполнение расчета возрастают сильнее.
Таким образом, мы видим, что все неэмпирические квантово-химические методы являются в большей или меньшей степени приближенными и их результаты очень сильно зависят от выбора базиса. Увеличение числа базисных функций обычно позволяет точнее передать распределение электронной плотности в молекулах и его изменение в ходе химических реакций. Вообще говоря, желательно расширять базис до выхода на хартри-фоковский предел. Однако сделать это удается лишь для простейших молекул. Для более сложных соединений приходится идти на компромисс между точностью расчета, с одной стороны, и затратами машинного времени (стоимостью расчета, возможностями ЭВМ) - с другой. Поэтому при изучении таких соединений приходится ограничиваться базисами среднего и даже минимального размеров, но тогда в расчет вносится значительный элемент произвола и теряется его универсальность, т.е. оказывается, что различные параметры молекул нужно рассчитывать в разных базисах.
Кроме того, использование приближения Хартри-Фока (метода ССП) также может вносить немалую ошибку в результаты расчета. В этом приближении вероятность найти электрон в некоторой точке пространства не зависит от местонахождения других электронов, распределение в пространстве которых задано одноэлектронными волновыми функциями. В результате двум электронам с одинаковыми спинами не запрещено занимать одну и ту же точку пространства. В действительности электроны с одинаковыми спинами стремятся избежать находиться не только в одной точке пространства, но даже близко друг от друга. Другими словами, электроны движутся зависимо, т.е. их движение носит коллективный характер. Пренебрежение этим эффектом, который принято называть электронной корреляцией, приводит к существенному завышению энергии взаимодействия электронов и, как следствие, завышению полной энергии молекулы.
Учет электронной корреляции является весьма трудоемкой задачей, требующей очень больших затрат машинного времени. Для этой цели в настоящее время чаще всего используют различные варианты метода конфигурационного взаимодействия, теории возмущений или метода электронных пар. В методе конфигурационного взаимодействия [1] многоэлектронную волновую функцию Ф ищут в виде линейной комбинации большого числа детерминантов Ψk:
Ф=∑CkΨk, (7)
где Ck— коэффициенты разложения; Ψk(где k≥1) — детерминанты, которые построены из одноэлектронных волновых функций основного состояния и отличаются от Ψo(волновой функции основного состояния, рассчитанной в приближении Хартри-Фока) тем, что один, два или более электронов перешли с занятых МО на вакантные.
Расчеты по теории возмущений основаны на предположении, что электронная корреляция относительно мало меняет волновую функцию основного состояния. В программе ГАУССИАН-80, которая наиболее широко в настоящее время применяется для неэмпирических квантово-химических расчетов, корреляционные поправки учитываются по теории возмущений Мёллера—Плезетта. В работах [22, 23] был предложен вариант метода МПДП с учетом электронной корреляции по теории возмущений Бриллюэна—Вигнера (КМПДП).
В методе электронных пар использовано формальное выражение, в котором корреляционная энергия записана как сумма корреляционных эффектов от каждой электронной пары [24]. Простейшим вариантом этого метода является приближение независимых электронных пар (в зарубежной литературе принято сокращенное обозначение IEPA). В данном приближении не учитывается взаимодействие электронных пар, поэтому уравнение для каждой электронной пары решается отдельно, а полная корреляционная энергия вычисляется как сумма корреляционных энергий всех электронных паp. IEPA является относительно дешевым (с точки зрения затрат машинного времени) методом учета корреляционных эффектов, но энергия электронной корреляции в этом приближении получается сильно завышенной.
В приближении связанных электронных пар (в зарубежной литературе принято сокращенное обозначение СЕРА) [25] учитывается взаимодействие электронных пар, поэтому уравнения можно решить только с помощью итерационной процедуры. Результаты получаются более точными, но затраты машинного времени на такой расчет необычайно велики. Так, расчет с учетом энергии электронной корреляции методом СЕРА для переходного состояния в реакции Сl- + СН3Сl = ClСН3 + Сl-c фиксированной геометрией потребовал свыше 10 ч на ЭВМ с быстродействием около 10 млн операций в секунду (для сравнения расчет для этого переходного состояния без учета энергии электронной корреляции на аналогичной ЭВМ требует 15—20 мин).
Во Введении мы дали краткое описание квантово-химических методов. Более детальное знакомство с ними практически не требуется для решения прикладных задач. Это связано с тем, что на основе анализа приближений, сделанных при разработке того или иного квантово-химического метода, как правило, не удается установить область его применения и очертить круг задач, которые можно решить с его помощью. К сожалению, многие квантово-химические методы, которые лучше обоснованы с теоретической точки зрения, на практике дают плохие результаты и поэтому не применяются, а более грубые модели с удачно подобранными параметрами широко используются. Это связано с тем, что в любом квантово-химическом методе сделано достаточно много различных приближений. В некоторых методах ошибки, к которым приводят эти приближения, частично компенсируют друг друга и в результате получается хорошее согласие с экспериментом. Сказать заранее, будет или не будет иметь место такая компенсация, нельзя, поэтому выяснить область применения и охарактеризовать точность каждого конкретного метода можно лишь на основе численного эксперимента и систематизации опубликованного расчетного материала. Знание этого вопроса (какие параметры молекулы каким методом следует вычислять и какие при этом могут получиться ошибки) отличает "опытного" квантового химика от "неопытного". В следующих разделах книги мы постараемся помочь нашему читателю стать "опытным" квантовым химиком.