Квантовая химия
КВАНТОВАЯ ХИМИЯ, раздел теоретич. химии, в к-ром строение и св-ва хим. соединений, их взаимод. и превращения в хим. р-циях рассматриваются на основе представлений и с помощью методов квантовой механики. Квантовая химия тесно связана с экспериментально установленными закономерностями в св-вах и поведении хим. соед., в т. ч. с закономерностями, описываемыми классич. теорией хим. строения. Начало развитию квантовой химии положили работы ряда исследователей, выполненные в период становления квантовой механики. В. Гейзенберг (1926) впервые провел расчет атома гелия; В. Гайтлер и Ф. Лондон (1927) на примере молекулы водорода дали квантовомех. интерпретацию ковалентной связи. Их подход нашел дальнейшее развитие в работах Дж. Слейтера (1931) и Л. Полинга (1931) и получил назв. валентных связей метод. В этот же период Ф. Хунд (1928), Р. Малликен (1928), Дж. Леннард-Джонс (1929) и Э. Хюккель (1930) заложили основы широко распространенного в настоящее время молекулярных орбиталей метода. Одновременно появились и основополагающие работы Д. Хартри (1927) и В. А. Фока (1930), создавших самосогласованного поля метод, а также работы Дж. Слейтера (1929-30) по мат. основам конфигурационного взаимодействия метода. X. Бете (1929) и Дж. Ван Флек (1932-35) разработали кристаллического поля теорию, развитие к-рой привело к созданию поля лигандов теории, нашедшей широкое применение в координац. химии. Общая схема квантовохим. подхода. Квантовохим. рассмотрение атомов, молекул и более сложных систем, свободных или находящихся во внеш. поле, не зависящем от времени, обычно начинается с решения стационарного ур-ния Шрёдингера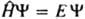 , где E и Y-полная энергия и волновая ф-ция системы,
, где E и Y-полная энергия и волновая ф-ция системы, 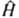 -оператор Гамильтона (гамильтониан) системы, представляющий собой сумму операторов кинетич. и потенц. энергии электронов и ядер, входящих в систему. Оператор кинетич. энергии равен:
-оператор Гамильтона (гамильтониан) системы, представляющий собой сумму операторов кинетич. и потенц. энергии электронов и ядер, входящих в систему. Оператор кинетич. энергии равен: 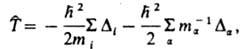
где индекс i нумерует электроны, индекс a-ядра, т и тa - массы электрона и ядра а соотв.,
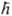 - постоянная Планка. В декартовых координатах Di и Da представляют собой сумму вторых частных производных по координатам электрона i и ядра a соотв., напр.:
- постоянная Планка. В декартовых координатах Di и Da представляют собой сумму вторых частных производных по координатам электрона i и ядра a соотв., напр.: 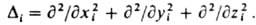
Оператор потенц. энергии равен сумме операторов кулоновского взаимод. частиц, к-рые зависят от расстояний rij между парами частиц, а также операторов взаимод. частиц с внеш. полем. Напр., для молекулы LiH (4 электтрона) в отсутствие внеш. поля гамильтониан выглядит след. образом:
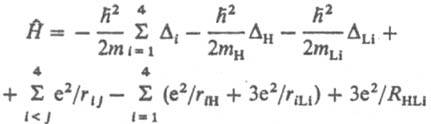
Ещё по теме
Квантовая химия — основы и применение
Квантово-химические методы в химии — применение и точность расчётов
Физическая химия — основы и методы исследования
Ядерная химия — открытия и методы исследования
Электронные представления в органической химии — от Беркенгейма до квантовой теории
Химическая физика — история и современные достижения
Химическая связь
Квантовая механика
Метод Хюккеля в квантовой химии
Химическая кинетика — основные понятия и законы
Квантовохимические расчёты в молекулярной спектроскопии
Теория химического строения органических соединений