Энергия. Пища
В древнеиндийском эпосе – «Махабхарате» – читаем: «Нет положения более бедственного,- сказал Шамбара,- чем то, когда ни сегодня, ни завтра утром не предвидится еды». И в другом месте: «О царь царей, пища – это жизнь, все живое обязано своим существованием прежде всего пище. Что мешает смерти победить жизнь? Пища!». Как видим, уже философы древней Индии, с характерной для них склонностью мыслить глобальными категориями, понимали потребность в пище гораздо глубже, чем просто как врожденный инстинкт. В их глазах это общебиологический закон, указывающий на необходимость непрерывно поддерживать извне хрупкое образование – жизнь, потому что жизнь – не стационарное состояние, а процесс. Современная медицина измеряе эту поддерживающую функцию пищи в условных калориях, подчеркивая тем самым преимущественно энергетическую роль пищи (а не структурную, иначе мы, наверное, говорили бы не о «калорийности» рационов, а, скажем, об их «молярности»). В этой главе мы коротко рассмотрим некоторые химические аспекты обмена энергией в живых системах, первоисточником которой для животных организмов служит пища (в обычном понимании этого слова), а для фотосинтезирующих – лучистая энергия («энергетическая пища» в чистом виде).
Давно стало прописной истиной, что жизнь, в любой ее форме, есть непрерывная борьба против второго начала термодинамики, против всемирной тенденции к спонтанному повышению суммарной энтропии. Общеизвестно также, как именно жизни удается добиться победы в этой, казалось бы, безнадежной схватке: биосфера перехватывает энергию солнечного излучения, запасает ее в форме энергии химических связей и затем расходует для обеспечения всей энергетики живых организмов и поддержания в них относительно низкого уровня энтропии до того, как эта энергия окончательно рассеется в виде тепла.
Углеводы стоят в начале и в конце этого грандиозного, непрерывно проходящего через биосферу потока энергии и энтропии: главными продуктами фотосинтеза являются гексозы, а главным источником энергии, удовлетворяющей повседневные потребности всех живых организмов, служит D-глюкоза.
Не будем разбирать ни реакции, протекающие при фотосинтезе, ни процессы, ведущие к утилизации энергии глюкозы: и те, и другие рассматриваются даже в современных школьных учебниках, не говоря уже об обширной научной литературе разной степени популярности. Однако с химических позиций небезынтересно задуматься о том, почему именно углеводам выпала ключевая роль во всей биоэнергетике.
Попытаемся мысленно сконструировать оптимальные структуры, пригодные для выполнения такой функции. В «техническое задание на проектирование» нам надо заложить два самых общих условия: материалом должны служить органические соединения, а главной средой для планируемых процессов – вода. Для более поздних этапов эволюции нужно прибавить и третье условие – существование окислительной (кислородной) атмосферы. Проектируемая система – биосфера – должна функционировать в замкнутом пространстве планеты, практически исключающем обмен веществом с внешней средой; единственное, что поступает в систему извне,- это солнечная энергия.
Поэтому наша система, рассчитанная на длительное существование, по материальному балансу должна быть замкнута в цикл: исходные и конечные продукты ее функционирования должны быть идентичны. Это производство – производство живых организмов – сверхмноготоннажное. В нем позволительно использовать только самое дешевое и массовое сырье. Главное, что нам нужно – это основные элементы – органогены: углерод, водород, кислород и азот. Их наиболее распространенные источники на современной Земле – углекислый газ, вода и молекулярный азот.
Три названных вещества представляют собой наиболее устойчивые формы существования четырех элементов в нынешних геологических условиях. Иными словами, им соответствует минимум потенциальной энергии. Следовательно, любые органические соединения, содержащие эти элементы или некоторые из них, будут обладать определенным запасом энергии по сравнению с «основным состоянием» (CO 2 +H 2 O+N 2 )и будут способны выполнять нужную нам функцию. Задача, однако, поставлена на оптимизацию: какие типы соединений будут справляться с этой функцией наилучшим образом?
Будем и дальше рассуждать как химики. Органические соединения содержат углерод в более восстановленной форме, чем в CO 2. В этом, в первую очередь, и заключается причина их энергоемкости в окислительной среде. Поэтому, оптимизируя решение по принципу наибольшей удельной емкости нашего аккумулятора, мы придем к наиболее восстановленным структурам – к предельным углеводородам. Вспомним, однако, что основная среда, согласно «техническому заданию»,- вода, в которой предельные углеводороды практически нерастворимы. Таким образом, и конструирование их молекул на стадии запасания энергии, и их распад на стадии утилизации энергии неизбежно включали бы гетерогенные реакции.
При прочих равных условиях гетерогенные реакции протекают резко замедленно по сравнению с гомогенными (это можно понять из самых общих соображений: гетерогенная реакция протекает на границе раздела фаз, т.е. в двухмерном пространстве, а гомогенная – в объеме, т.е. в трех измерениях). Поэтому биоэнергетика, построенная на предельных углеводородах, была бы в целом медленной: система была бы способна запасать много энергии, но обладала бы низкой мощностью. Это привело бы к большому замедлению всех жизненных процессов и, как следствие, к тому, что организмы оказались бы в гораздо большей степени подвержены воздействию неблагоприятных изменений внешней среды. Вывод: надо ориентироваться на более гидрофильные, растворимые в воде соединения, и при этом не слишком проиграть в энергоемкости по сравнению с углеводородами.
Повысить гидрофильность органической молекулы можно путем введения в нее полярных групп. Для создания таких групп пригодны два других имеющихся в нашем распоряжении элемента-органогена: кислород и азот.
При помощи каждого из них можно построить по три основных типа полярных группировок: карбоксильную (-COOH), карбонильную (=С=О) и спиртовую ( º С-OH) на основе кислорода, и амидиновую (-С(-NH 2 )=NH), имидную (=С=NH) и аминогруппу ( º С-NH 2 ) на основе азота.
Что выбрать: кислород или азот? Предпочтение следует отдать кислороду. И вот по каким соображениям. Все три азотные группировки в водной среде в большей или меньшей степени склонны к гидролизу до соответствующих кислородных функций. Поэтому системы на их основе будут потенциально нестабильны, что нежелательно для столь фундаментальной системы, как биоэнергетика. Кроме того, все эти группы (особенно аминогруппа и тем более амидиновая) обладают основным характером. Поэтому рН растворов таких соединений будет сильно отклоняться в щелочную область от рН природной воды (около 7), что создаст для организмов с такой энергетикой дополнительные неудобства.
Из трех кислородных функций логично выбрать спиртовую, так как она соответствует наиболее низкой степени окисления углерода, а значит, наиболее высокому запасу энергии. Как показывает опыт органической химии, для того чтобы спирты хорошо растворялись в воде, идеально «вписывались» в ее структуру, нужно у каждого или почти у каждого углеродного атома иметь по одной гидроксильной группе. Так мы приходим к многоатомным спиртам типа HO-CH 2 -(CHOH) n -CH 2 -OH.
Создание таких молекул из CO 2 и воды (на стадии запасания энергии) и их расщепление (на стадии утилизации энергии) обязательно требуют образования и расщепления C-C-связей. Оба эти процесса в алифатическом ряду обычно сопряжены с преодолением значительных потенциальных барьеров, что, разумеется, отрицательно сказывается на скоростях соответствующих реакций. Конечно, можно положиться на всемогущество биокатализаторов – ферментов, но все-таки лучше заранее облегчить им работу. Для этого надо подобрать какую-нибудь реакцию, которая легко протекает в водной среде и обратимо ведет к образованию С-С-связей в полиоксисоединениях.
Такая реакция есть: это альдольная конденсация α -оксикарбонильных соединений и обратная ей реакция – ретроальдольный распад. Простейший ее пример – конденсация гликолевого альдегида по схеме:
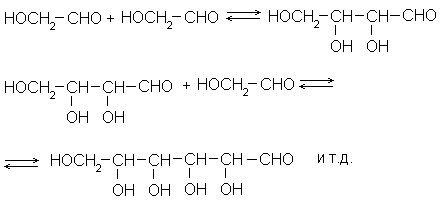
Продуктами такой реакции являются полиоксиальдегиды. Для перехода к ним от многоатомных спиртов, к которым мы пришли на предыдущих этапах рассуждений, в структуру последних нужно внести лишь минимальные изменения: одну альдегидную группу. При этом мы проиграли в запасе энергии лишь минимально (один атом углерода оказался более окисленным, чем другие), но зато приобрели реакционную подвижность С-С-связей углеродного скелета, т.е. наиболее консервативного элемента структуры.
Теперь надо решить, какова должна быть оптимальная длина углеродной цепи в наших соединениях. Но здесь положение сущственно меняется. Вопрос о длине цепи природа материала решает за нас. Действительно, мы помним, что оксикарбонильные соединения легко и самопроизвольно замыкаются в кислородные гетероциклы, из которых наиболее устойчив шестичленный – пиранозный – цикл. При последовательном росте цепи полиоксиальдегида возможность замыкания пиранозного цикла впервые появляется у C 5 -оксиальдегидов, а если цепь наращивается сразу на два углеродных атома, как при конденсации гликолевого альдегида, то у соединений C 6. После замыкания пиранозного цикла карбонильная группа в молекуле исчезает (по крайней мере в явном виде) и дальнейший рост или распад цепи по схеме альдольной конденсации или ретроальдольного распада становится затруднительным. Так что подобные реакции автоматически останавливаются на молекулах C 5 и C 6, т.е. на моносахаридах альдопентозах и альдогексозах.
Теперь можно обратить внимание на то, что в полиоксиальдегидах все углеродные атомы, кроме концевых, являются асимметрическими центрами. Придавая им определенные конфигурации, можно “нагружать” эти молекулы дополнительной информацией (т.е. “отрицательной энтропией”), что даст живым организмам добавочные преимущества в их противодействии второму началу термодинамики.
Пойдем дальше. Задумаемся над вопросом об оптимальной конфигурации этих центров. Из данных конформационного анализа легко заключить, что в ряду стереоизомерных полиоксиальдегидов наиболее устойчивым будет тот, у которого все заместители при пиранозном цикле экваториальны. Такая структура возникает в ряду альдопентоз для ксило-, а в ряду альдогексоз – для глюко-конфигурации. А универсальной единицей хранения энергии в реальных живых системах является D-глюкоза! Так, исходя из самых общих предпосылок и законов логики органической химии, приходим к тому же, к чему привела эволюцию стихийная логика природы.
Зададимся, наконец, вопросом, почему основным хранилищем энергии оказались углеводы, а не АТФ (аденозинтрифосфат), как известно, служащий единой «разменной монетой» в энергетике всех живых систем. Не лучше ли было бы и запасать, и хранить, и расходовать энергию в виде такого универсального вещества и не прибегать к помощи углеводов? Оказывается, что нет.
АТФ, как разменная монета удобна для покрытия мелких расходов и весьма мало пригодна для хранения сбережений. Действительно, при полном окислении глюкозы в клетке образуется 11 молекул АТФ (или его энергетических эквивалентов). Молекулярная масса глюкозы – 180, а натриевой формы АТФ – 573 дальтона. Таким образом, удельные энергоемкости глюкозы и АТФ относятся как 35:1. Так что глюкозы – гораздо более компактное хранилище энергии.
Существует способ еще несколько повысить удельню энергоемкость углеводов. Это поликонденсация моносахаридов в полисахариды. Вот по каким причинам организмы прибегают к этому способу.
Глюкоза чрезвычайно легко растворима в воде. Мы видим в этом важное преимущество моносахаридов как хранилищ энергии перед углеводородами. Но это преимущество, однако, не абсолютно и превращается в недостаток при необходимости депонирования значительных энергетических ресурсов. В самом деле, для этого в клетке должен был бы содержаться весьма концентрированный раствор глюкозы, что невыгодно и по физико-химическим, и по биохимическим причинам. Поскольку глюкоза – низкомолекулярное соединение, это было бы связано со значительным повышением осмотического давления в клетке и с тенденцией легкой диффузии глюкозы наружу. Для противодействия подобным нежелательным факторам потребовалось бы создание сложных специализированных механизмов. С другой стороны, глюкоза представляет собой субстрат многих ферментативных реакций в клетке, и для ограничения ее чрезмерного расходования во время хранения клетка должна была бы располагать множественными и разнородными системами. Напротив, создание запасов энергии в форме специализированных полисахаридов (так называемые резервные полисахариды) обеспечивает живым организмам целый ряд преимуществ.
Поликонденсация моносахаридов в полисахариды сопряжена с отщеплением воды и, следовательно, со снижением молекулярной массы, отнесенной к потенциальной моносахаридной единице (для глюкозы – на 10%). Некоторая дополнительная энергия запасается в форме энергии образующихся гликозидных связей (гидролиз гликозидов – слабоэкзотермическая реакция). Резервные полисахариды высокомолекулярны, а большинство из них нерастворимо в воде при физиологических условиях. Так что все отрицательные эффекты хранения в клетках больших количеств свободной глюкозы снимаются.
Для утилизации энергии резервных полисахаридов путем их расщепления до моносахаридов нужно ограниченное число специфических ферментов – полисахаридаз. Это обеспечивает организмам возможность эффективного управления поступлением свободной глюкозы, т.е. в конечном счете расходованием запасенной энергии, как путем активации или угнетения этих ферментных систем, так и при помощи включения или блокирования биосинтеза соответствующих ферментов.
В животных организмах функцию резервного полисахарида выполняет гликоген, в большинстве растений – крахмал (амилоза+амилопектин), в бурых водорослях – ламинарин, в дрожжах и бактериях – декстраны. (Заметим, что все эти полисахариды построены только из остатков D-глюкопиранозы.) Высшие растения накапливают крахмал в особенно больших количествах в органах, связанных с воспроизведением вида, где необходимо создавать значительные энергетические ресурсы для обеспечения развития зародыша: в семенах при половом размножении и корневищах – при вегетативном (их клетки часто оказываются буквально «битком набиты» крахмальными гранулами). Именно эти части растений составляю основу растительной пищи и главный источник пищевой энергии для современного человека (семена злаковых, клубни картофеля и т.п.). В организмах животных гликоген накапливается в первую очередь в клетках печени и мышц. Рассмотрим немного подробнее функцию гликогена печени.
В печени гликоген играет роль буфера глюкозы, циркулирующей в крови и являющейся главным энергетическим ресурсом всех клеток организма. Концентрация глюкозы в плазме крови должна поддерживаться постоянной: падение ее ниже нормы приводит к голоданию клеток и оказывается гибельным для тех из них, которые неспособны создавать собственные энергетические резервы (каковы, например, клетки головного мозга), а превышение ведет к резким биохимическим сдвигам в клетках, и также особенно опасно для клеток мозга. Между тем и расходование глюкозы плазмы, и ее поступление подвержены резким колебаниям. Например, при переходе от покоя к активной деятельности убыль глюкозы скачкообразно возрастает, а при переваривании пищи, особенно углеводной, в кровь быстро поступают значительные количества глюкозы. Таким образом, понятно, что организм должен располагать быстродействующими и легко управляемыми механизмами биосинтеза гликогена (депонирование избыточной глюкозы плазмы) и его расщепления (компенсация энергетических затрат). На примере расщепления гликогена удобно проследить связь его структуры с выполняемой функцией.
Гликоген построен из остатков α -D-глюкопиранозы и имеет высоко разветвленную структуру (см. с. 144). Связь остатков в цепях 1 à 4, в точках разветвлений – 1 à 6. Количественные параметры, характеризующие структуру гликогена варьируют, в зависимости от его источника (вида животного, природы ткани). В типичных случаях внешние неразветвленные цепи (А) содержат шесть-десять моносахаридных остатков, а во внутренних цепях (В и С) между разветвлениями находится два-четыре остатка глюкозы. Молекулярные массы гликогенов широко варьируюся и могут достигать десятков миллионов дальтон. (Это весьма значительная величина даже для биополимеров; она превышает, например, массу многих вирусных частиц.) В отличие от большинства других резервных полисахаридов гликоген хорошо растворим в воде.
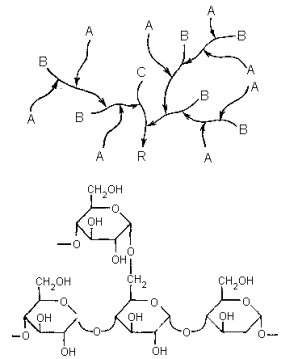
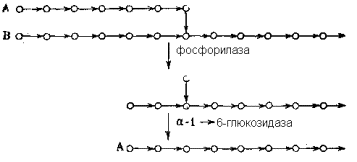
где I-общая схема структуры (А-концевые неразветвленные цепи, В-цепи, несущие разветвления, С-единственная в молекуле цепь с восстанавливающим концом, R-восстанавливающий конец);II-детальная структура участка, примыкающего к узлу разветвления;III-схема, иллюстрирующая первые две стадии ферментативного расщепления гликогена (кружки символизируют остатки α -D-глюкопиранозы, горизонтальные стрелки – 1 à 4-связи, вертикальные – 1 à 6-связи).
Расщепление гликогена в печени катализируется двумя ферментами: гликоген-фосфорилазой и α -1,6-глюкозидазой. Оба фермента высоко специфичны к структуре отщепляемого остатка (отщепляют только концевой остаток α -D-глюкопиранозы), и к типу разрываемой связи (первый расщепляет только 1 à 4-связи, второй – только 1 à 6), и к структуре цепи, прилегающей к разрываемой связи со стороны восстанавливающего конца.
Особенно прихотлива специфичность последнего типа у фосфорилазы. Она отщепляет один за другим все 1 à 4-связанные остатки неразветвленных цепей (типа А) и оставляет неотщепленным последний остаток глюкозы, связанный с другой цепью 1 à 6-связью в точке ветвления. В цепях такого же типа В расщепление происходит аналогично, но останавливается за несколько 1 à 4-связанных остатков до точки разветвлкения. Далее на частично расщепленный гликоген действует α -1,6-глюкозидаза, которая гидролизует 1 à 6-связь одиночного глюкозного остатка, сохранившегося в точке ветвления после действия фосфорилазы. В результате остаток цепи типа B превращается в цепь типа А, после чего включается следующий цикл реакций, катализируемой фосфорилазой, пока этот фермент вновь не «споткнется» на очередных разветвлениях, после чего вновь вступает в «игру» глюкозидаза и т.д.
Из описанной схемы ферментативного расщепления гликогена легко видеть, насколько целесообразно построена молекула гликогена в свете его основной функции. В самом деле, благодаря исключительно высокой разветвленности цепей гликогена каждая его молекула содержит большое число невосстанавливающих концов цепей (порядка десятков тысяч), так что одна молекула полисахарида может подвергаться атаке одновременно во многих местах. Это обстоятельство обеспечивает чрезвычайно высокую скорость расщепления и, следовательно, возможность почти мгновенной мобилизации заключенных в гликогене энергетических ресурсов.
Биологический смысл субстратной специфичности расщепляющих гликоген ферментов легко понять: «ключ» от энергетического «сейфа» – молекулы гликогена – должен идеально подходить к «замку», особенно если вспомнить, что это хранилище должно безотказно открываться в аварийных ситуациях (например, при стрессах). И в то же время, эти ферменты не должны действовать ни на какие другие углеводные структуры, иначе при их включении клетки печени не только выдадут нужную организму глюкозу, но и быстро разрушат сами себя. Однако и замок должен точно соответствовать ключу. Иными словами (и тут мы вплотную подходим к вопросу о специфичности полисахаридных структур), вариации структуры гликогена допустимы лишь в таких пределах, в которых он сохраняет способность быть хорошим субстратом для расщепляющих его ферментов печени. А это, как мы видели, очень узкие пределы, не допускающие, например, ни наличия в молекуле фуранозных звеньев, ни изменения конфигурации гликозидных связей, ни наличия межмономерных связей, отличных о 1 à 4 и 1 à 6, ни замены части остатков глюкозы на остатки других моносахаридов и т.д. На любой из таких аномалий, даже если их содержание в молекуле незначительно, ферментативное расщепление гликогена приостановится, и вся остальная, и весьма значительная, часть полисахаридной молекулы не сможет быть вовлечена в обмен.
Еще несколько слов о специфичности полисахаридов. Когда говорят о субстратной специфичности ферментов, обычно (особенно в научно-популярной литературе) делают акцент исключительно на способности фермента отличать свой субстрат от огромного числа других веществ, описывают фермент как изумительную распознающую машину, идеально приспособленную для выполнения своей функции.
При таком взгляде явно или неявно подразумевается пассивность субстрата в ферментативном акте, ему отводится роль инертного материала, над которым производится некая операция. Между тем современная концепция ферментативного катализа (концепция взаимно-индуцированного соответствия) отводит обоим компонентам взаимодействия фермент-субстрат равноправные активные роли. Суть ее в том, что при образовании фермент-субстратного комплекса происходит одновременное изменение конформации и субстрата, и фермента; это дает в итоге идеальную подгонку молекул обоих участников одна к другой.
Образно говоря, это не механическое наложение двух твердых предметов с комплементарной конфигурацией («ключа» и «замка»), а скорее, взаимная настройка гибких объектов. В каждом из участников такого взаимодействия, взятом изолированно, предсуществует не готовая матрица, соответствующая структуре другого участника, а лишь способность создавать такую матрицу под влиянием второго компонента.
Таким образом, мы можем уверенно говорить о высокой специфичности углеводов по отношению к катализирующим их реакции ферментам, об их способности узнавать «свои» ферменты среди множества других веществ с таким же правом, с каким мы говорим о субстратной специфичности этих ферментов.