1.3. РАСЧЕТ ИНДЕКСОВ РЕАКЦИОННОЙ СПОСОБНОСТИ
Для некоторых реакций наблюдается хорошая корреляция между выходами конечных продуктов и так называемыми индексами реакционной способности. В качестве индексов обычно используют заряды на атомах, порядки связей, энергии граничных МО, квадраты коэффициентов разложения граничных МО по базису АО (парциальные электронные плотности) и т.д. Здесь термином "граничные МО" обозначены верхняя занятая и нижняя вакантная МО соединения. Наиболее часто пользуются зарядами на атомах и параметрами граничных МО, поэтому мы остановимся только на этих индексах реакционной способности.
Почему же имеют место корреляции между индексами реакционной способности и выходом конечных продуктов реакций? Вообще говоря, выходы конечных продуктов реакции определяются свободными энергиями активации, а индексы реакционной способности характеризуют энергию межмолекулярных взаимодействий для достаточно удаленных друг от друга реагентов. Однако для некоторых реакций эти параметры (свободные энергии активации и энергии межмолекулярных взаимодействий) количественно связаны друг с другом, т.e. с увеличением энергии взаимодействия между реагентами свободная энергия активации уменьшается.
Энергию межмолекулярного взаимодействия при сближении реагентов можно условно разбить на вклады трех типов: кулоновские, орбитальные и стерические. Энергия кулоновского взаимодействия зависит от распределения электронной плотности или от зарядов на атомах реагентов. Поэтому для некоторых реакций удается найти корреляцию между зарядами на атомах и выходом конечных продуктов реакции. Так, нуклеофильные реагенты (атакующий центр заряжен отрицательно) присоединяются преимущественно к атомам, на которых локализованы большие положительные заряды, а электрофильные (атакующий центр заряжен положительно), наоборот, — к атомам, на которых локализованы большие отрицательные заряды.
Корреляции между выходом конечных продуктов реакции и зарядами на атомах широко используются химиками-синтетиками для объяснения экспериментальных данных, и здесь все более или менее ясно. Отметим только, что понятие заряда на атоме имеет условный характер. На атоме в действительности локализован лишь заряд ядра, электроны внутренних оболочек находятся вблизи ядер, а валентные электроны локализованы между атомами. Обычно при вычислении заряда на атоме в квантовой химии пользуются анализом электронных заселенностей, предложенным Малликеном [31]. В этом приближении заряд qA на атоме А вычисляется по следующей формуле:
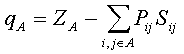
Здесь сумма берется по всем орбиталям i и j атома A; ZA — заряд ядра;
Рij- матрица зарядов и
порядков связей; Sij- матрица интегралов
перекрывания. В полуэмпирических методах обычно пользуются упрощенной формулой 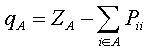
Величины зарядов на атомах, вычисленные в этом приближении, в неэмпирических расчетах очень сильно зависят от выбора базиса, а в полуэмпирических — от выбора метода. Заряды на атомах, вычисленные в разных базисах (неэмпирические расчеты) и разными методами (полуэмпирические расчеты), могут различаться в 1,5—2 раза, но качественные результаты (знак и относительная величина заряда) обычно остаются одинаковыми. В неэмпирических расчетах заряды на атомах при расширении базиса обычно увеличиваются по абсолютной величине.
Формально заряды на атомах в приближении Малликена можно вычислить в любом базисе, но при включении в него сильно диффузных орбиталей результаты становятся сомнительными, так как атому приписывается электронная плотность, которая в действительности от него сильно удалена. В частности, не следует использовать анализ заселенностей по Малликену при включении в базис поляризационных орбиталей (расчеты в базисах 6-31ГФ* и 6-31ГФ**) и диффузных s- и p-орбиталей (расчеты в базисе 3-21+ГФ). Отметим также, что заряды на атомах, вычисленные методом ППДП/2, обычно близки к результатам неэмпирического расчета в минимальном базисе, а методы МЧПДП/3 и МПДП дают приблизительно такое же распределение электронной плотности, как неэмпирические расчеты в валентно-расщепленных базисах.
При использовании зарядов на атомах для изучения реакций, которые идут в одну стадию или у которых первая стадия определяет направление и выход конечных продуктов, достаточно рассчитать электронную структуру исходных реагентов и провести корреляцию между вычисленными зарядами на атомах и направлением реакции. Такие работы хорошо известны. В частности, так обычно объясняют влияние заместителей на направление реакций нуклеофильного и электрофильного замещения ароматических соединений. Однако для большинства реакций подобные корреляции провести не удается. Иногда можно найти корреляцию между электронным строением интермедиата, который образуется на одной из элементарных стадий, и выходами конечных продуктов реакции. В этом случае квантово-химические расчеты приходится проводить для различных метастабильных промежуточных продуктов и отбирать из них интермедиаты, электронная структура которых позволяет объяснить экспериментально наблюдаемое направление реакции. Так, на основе расчета электронной структуры тиофеновых α,β-непредельных кетонов в работах [32, 33] не удалось объяснить направление реакции нитрования в сильно кислых средах, поэтому пришлось провести расчеты для протонированных форм, установить, какие интермедиаты, образующиеся при протонировании, являются наиболее устойчивыми, и лишь на основе анализа их электронной структуры удалось найти корреляцию между направлением нитрования и зарядами на атомах. Однако для большинства реакций подобные корреляции найти не удается. Под термином "орбитальное взаимодействие" понимают взаимодействие между молекулярными орбиталями реагентов при их сближении. Это взаимодействие имеет квантово-механическое происхождение. По своей природе оно близко к хорошо известному химикам эффекту сопряжения. Рассмотрим механизм орбитального взаимодействия на качественном уровне. При сближении реагентов Х и Y между их орбиталями φX и φYвозникает перекрывание и, как следует из квантовой теории, разность энергий E(φX)-E(φY) увеличивается по абсолютной величине.
Пусть E(φX)>, тогда взаимодействие между φX и φYприведет к уменьшению E(φX) и увеличению E(φY) на одинаковые значения (рис. 1.2). Если φX и φY— занятые орбитали соединений Х и Y, то полная энергия системы X...Y не изменится (рис. 1.2,а), но если φX— занятая орбиталь, а φY— вакантная, то произойдет понижение полной энергии реагентов (рис. 1.2,б), другими словами, между реагентами появится орбитальное взаимодействие. Таким образом, при сближении реагентов Х и Y взаимодействие занятых МО φXi и вакантных МО φ*Ykприводит к понижению положения занятых МО φXi на шкале энергий и к стабилизации системы X...Y. К аналогичному результату приводит взаимодействие занятых МО φYkи вакантных МО φ*Xi.
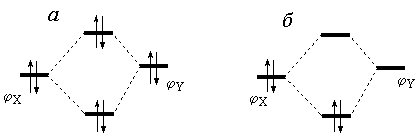
Рис. 1.2. Орбитальное взаимодействие реагентов Х и У
Энергию орбитального взаимодействия можно оценить во втором порядке теории возмущений [2]:
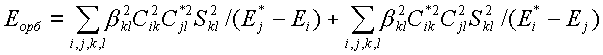
Здесь сумма по i берется по всем занятым (первое слагаемое) и всем вакантным (второе слагаемое) МО X, сумма по jберется по всем вакантным (первое слагаемое) и занятым (второе слагаемое) МО Y, сумма по k берется по всем АО X, по l — по всем АО Y; βkl— коэффициент, имеющий размерность энергии; Сik и Сjl— коэффициенты разложения МО Х и Y по базисным АО; Sij — интеграл перекрывания; Е*i, E*j, Еi и Ej — энергии МО φ*Xi, φ*Yj, φXiи φYj.
Стабилизация системы X...Y за счет орбитального взаимодействия любых пар МО обратно пропорциональна разности их энергий, т.е. чем дальше друг от друга лежат орбитали на шкале энергий, тем слабее они взаимодействуют. Поэтому на практике обычно пользуются приближением граничных орбиталей, т.е. учитывают взаимодействие лишь между двумя МО, для которых разность энергий минимальна. В этом приближении энергия орбитального взаимодействия зависит от энергий граничных МО и от коэффициентов разложения этих МО по базису АО. Любую из этих величин можно использовать в качестве индекса реакционной способности, но наиболее часто пользуются разностью энергий граничных МО. Так, в работах [34, 35] при изучении реакции Дильса—Альдера циклических диенов (циклопентадиена, гексахлорциклопентадиена и тетрахлор-1,2-бензохинона) с диенофилами (монозамещенными ацетиленами, сопряженными аминами, сопряженными диенами и триенами) была обнаружена корреляция между выходами конечных продуктов реакции и положением граничных МО на шкале энергий. Этот результат свидетельствует, что для данной реакции определяющую роль играет орбитальное взаимодействие.
Индексы реакционной способности весьма широко применяются в прикладной квантовой химии, однако с их помощью можно решать лишь весьма ограниченный круг вопросов. В большинстве случаев они не позволяют определить ни направление, ни относительную скорость реакции, поэтому для изучения реакционной способности органических соединений приходится применять более сложные методики (расчеты тепловых эффектов и поверхностей потенциальной энергии). В качестве примера рассмотрим результаты работы [36], в которой был изучен механизм присоединения СН-кислот типа XCH2CО2Et (X = CO2Et, COMe, CN) к α,β-непредельным альдегидам в условиях межфазного катализа. Из эксперимента было известно, что эта реакция может идти по двум направлениям: карбанион, генерированный из СН-кислоты, может присоединяться к карбонильному атому углерода и к β-атому углерода связи С=С.
Предполагалось, что в первом случае направление реакции определяется электростатическим взаимодействием (зарядовый контроль), а в во втором — орбитальным (орбитальный контроль). Влияние заместителей на направление присоединения при этом объясняли изменением относительной величины электростатического и орбитального взаимодействий. Однако результаты квантово-химических расчетов [36] показали, что это не так. Оказалось, что такие заместители, как хлор, метильная и фенильная группы, практически не меняют относительную величину орбитального и электростатического взаимодействий, хотя и меняют направление присоединения.
Все попытки объяснить влияние заместителей на направление реакции с помощью статических индексов реакционной способности окончились безрезультатно. Дальнейшее исследование показало, что в данном случае на направление присоединения основное влияние оказывает стерический эффект. Мы специально привели этот пример, так как при изучении присоединения заряженного реагента к ненасыщенному атому углерода, как правило, удается найти корреляцию между направлением присоединения и индексами реакционной способности (обычно π- или π+σ-электронными зарядами на атомах), но даже и здесь, как видим, есть исключения. Для других классов органических реакций область применения индексов реакционной способности уменьшается.
В заключение этого раздела подчеркнем еще одно очень важное обстоятельство. При поиске корреляций между индексами реакционной способности и выходами продуктов реакции необходимо располагать достаточно большим материалом для сравнения. Если имеются данные лишь для 5—7 родственных соединений, то статистическая вероятность сделать ошибочное заключение будет очень велика. Кроме того, при поиске корреляций между результатами квантовохимических расчетов для газофазных моделей и данными эксперимента, полученными в растворе, необходимо помнить, что растворитель очень сильно меняет электронную структуру ионов, при этом наиболее значительно меняются энергии МО.