3.1. СИЛОВЫЕ ПОСТОЯННЫЕ ХИМИЧЕСКИХ СВЯЗЕЙ И ЧАСТОТЫ ВНУТРИМОЛЕКУЛЯРНЫХ КОЛЕБАНИЙ
Силовая постоянная химической связи K(i), равная второй производной полной энергии молекулы по удлинению данной связи qi, (∂2E/∂qi2), является, пожалуй, основной характеристикой механических свойств связи. Для одноименных связей в разных молекулах сходного типа она варьируется в небольших пределах. Известны полуколичественные корреляции ее с длиной и порядком связи, а также некоторые взаимосвязи с энергией диссоциации.
Химическая связь в многоатомной молекуле, строго говоря, не может быть охарактеризована одним силовым коэффициентом. Для этой цели требуется введение матрицы коэффициентов, включая те, которые связаны с взаимодействием данной связи и всех других связей, т.е. недиагональные элементы [132,133]: K(i,j) = ∂2E/∂qi∂gj. Последние определяют появление сил, действующих в j-x связях, в результате растяжения i-й связи. Недиагональные элементы, как правило, невелики, но для решения некоторых задач они имеют первостепенное значение.
Если располагать данными о структурной формуле соединения и необходимым набором силовых постоянных, то для изучаемой конформации можно довольно просто вычислить частоты всех нормальных колебаний атомов ω, которые проявляются в ИК- и КР-спектрах, электронно-колебательных спектрах поглощения и вторичного излучения. Можно также определить формы нормальных колебаний и охарактеризовать силовое поле в как угодно деформированной молекуле. Эти возможности лимитируются недостаточностью сведений о самих силовых постоянных, которые особенно ограниченны для электронно-возбужденных состояний.
Общее количество информации, требующейся для точного расчета частот ω сложных молекул, весьма велико, и имеющиеся отрывочные данные приходится дополнять, используя аналогии, а иногда и весьма сомнительные допущения. В этих условиях привлечение дополнительных данных представляется очень важным. Квантовая химия занимает здесь особое место, так как позволяет в принципе не только оценить любые силовые постоянные как для основного, так и для электронно-возбужденных состояний, но и выяснить для отдельных коэффициентов их происхождение и связь со строением электронной оболочки, чему, к сожалению, пока не уделялось достаточного внимания.
Известно, что решение обратной задачи, т.е. вычисление силовых коэффициентов по известным из эксперимента колебательным частотам, требует большой и трудной подготовительной работы по синтезу изотопных моделей. Во многих случаях решение неоднозначно и выбор между наборами коэффициентов, удовлетворяющих исходной системе уравнений, основывается нередко на интуиции. Привлечение методов квантовой химии дает в распоряжение исследователя независимые критерии для такого выбора. К сказанному можно добавить, что квантовохимический расчет силовых постоянных не связан с какими-либо дополнительными трудностями и может быть выполнен обычными методами, описанными во введении.
Для расчета силовых постоянных довольно широко применяют как полуэмпирические, так и неэмпирические методы квантовой химии. В любом случае сначала оптимизируют геометрию, т.е. определяют наиболее устойчивую конформацию, отвечающую минимуму полной энергии; затем вычисляют вторые производные полной энергии по декартовым или естественным координатам (силовые постоянные), а при необходимости - кубичные и биквадратичные члены.
Выбор базиса существенно сказывается на результатах неэмпирических расчетов силовых постоянных (в особенности недиагональных элементов). Так, при использовании минимального слейтеровского базиса согласие с экспериментом получается весьма посредственное. Точность расчета существенно повышается при переходе к валентно-расщепленному базису (4-31ГФ, 4-21ГФ или 3-21ГФ), но его дальнейшее расширение уже малоэффективно. При выходе на так называемый хартри-фоковский предел силовые постоянные оказываются завышенными на 10 - 20% [134], при этом точность расчета недиагональных элементов намного ниже. Так, для молекулы СО2 значение К(С—О, С—О/) получилось завышенным почти в 2 раза [135]. Учет электронной корреляции улучшает результаты [136, 137], однако для многоатомных молекул он практически недоступен.
Для полуэмпирических методов характерно относительное занижение частот деформационных колебаний по сравнению с валентными. При этом в методе ППДП/2 без введения поправок значения колебательных частот для деформационных мод хорошо согласуются с экспериментом, а данные для валентных колебаний получаются сильно завышенными. В методе МЧПДП последние, напротив, близки к опытным данным, но частоты деформационных колебаний занижены. Методы МЧПДП/3 и МПДП дают результаты промежуточного типа: значения частот для валентных колебаний получаются немного завышенными, а для деформационных - заниженными. Ошибки при использовании методов МЧПДП/3 и МПДП получаются примерно такие же, как в неэмпирических расчетах без учета электронной корреляции (в среднем около 200 см-1).
В связи с тем, что ошибки в большинстве случаев носят систематический характер, их удается значительно уменьшить введением эмпирически подобранных масштабных корректирующих множителей для определенных типов силовых постоянных (например, для связей С—С, углов С—С—Н и т.д.) или инкрементов, которые прибавляются к рассчитанным частотам.
Для расчетов методами МЧПДП/3 и МПДП Дьюар и Форд [138, 139] подобрали систему инкрементов, специфичных для валентных, деформационных и торсионных колебаний определенных атомных групп или связей; на очень большом числе примеров продемонстрирована удовлетворительная точность результатов. Однако многообразие форм нормальных колебаний, которые в сложной молекуле включают колебания многих связей и углов, и невозможность четкого разграничения валентных и деформационных мод ограничивают перспективы корректирования частот по типам колебаний.
Более логичным представляется корректирование значений силовых постоянных, и на этом пути достигнуты положительные результаты. В настоящее время используется несколько методик подбора корректирующих множителей. Они различаются способом корректировки недиагональных силовых коэффициентов. В' работе [140] предложено корректировать только диагональные силовые постоянные, а недиагональные оставить без изменений. Этот подход использован в работе [141], в которой методом МЧПДП/3 рассчитаны частоты нормальных колебаний этана, пропана, этилена, бутена-2 и 1,3-бутадиена. Для улучшения количественного согласия с экспериментом были уменьшены силовые постоянные валентных связей на 30%, увеличены силовые постоянные валентных и торсионных углов на 20%, а недиагональные коэффициенты оставлены без изменений. Частоты нормальных колебаний исследованных соединений, вычисленные с такой корректировкой силового поля, отличаются от экспериментальных значений в среднем на 50см-1.
Блом и Алтона [142 - 144] использовали один общий корректирующий множитель для всех недиагональных силовых постоянных. Так, при неэмпирическом расчете в базисе 4-31ГФ колебательных спектров этилена ими использовано пять корректирующих множителей порядка 0,75 - 1,0. Из них четыре - для диагональных силовых постоянных и один - для всех недиагональных членов. При этом среднее расхождение с наблюдаемыми на опыте 12 колебательными частотами уменьшилось более чем в 10 раз и составило около 7 см-1 [142]. Подобные результаты получены и для этана, пропана, пропена и диметилового эфира, но корректирующие коэффициенты подбирались для каждого соединения индивидуально [143, 144]. Отметим, что простое определение частот по набору коэффициентов, полученных из опыта для других молекул сходного типа, дает при отсутствии сопряжения примерно такую же точность.
В работах Пулея с сотрудниками
[145, 146] предложено подбирать значения корректирующих множителей только для
диагональных членов, а корректирующие множители для недиагональных коэффициентов
вычислять как среднее геометрическое из соответствующих диагональных величин,
т.е. если Сi и Сj-
корректирующие множители для K(i)
и K(j), то корректирующий
множитель для K(i,j)
- 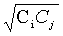 . В настоящее время методика Пулея используется наиболее
широко.
. В настоящее время методика Пулея используется наиболее
широко.
Первые работы с использованием корректирования силового поля молекулы по методике Пулея выполнены полуэмпирическими методами. Так, было показано [146], что при расчете колебательных частот углеводородов методом ППДП/2 следует ввести следующие корректирующие множители: 0,38 для силовых постоянных валентных связей; 0,80 для силовых постоянных валентных углов; 0,62 для силовых постоянных углов выхода из плоскости молекулы. Такие же поправки были использованы для глиоксаля и акролеина [147]. Набор масштабных множителей для органических соединений, содержащих атомы N и О, рекомендован и опробован в работе [148], при этом среднеквадратичная ошибка в ряду весьма сложных триазолов не превышала 30 см-1.
Были рассчитаны [149] частоты нормальных колебаний 4Н-пиран-4-она методами ППДП/2 и МЧПДП/3. Для каждого метода подобран свой набор корректирующих множителей; при этом предполагалось, что их значения для связей и углов, образованных разными атомами, различаются. В случае метода ППДП/2 значения вычисленных корректирующих множителей для валентных связей лежат в области 0,25 - 0,45; для валентных углов в плоскости молекулы — 0,83 - 1,20; для углов выхода из плоскости молекулы — 0,65 - 0,80. При использовании метода МЧПДП/3 значения подобранных корректирующих множителей для валентных связей лежат в области 0,70 - 0,80; для валентных углов — 1,15 - 1,30; для углов выхода из плоскости молекулы — 1,20 - 1,30. Средние отклонения рассчитанных колебательных частот от экспериментальных величин в случае метода ППДП/2: для плоских колебаний 20 см-1, для неплоских колебаний 33 см-1; в случае метода МЧПДП/3: для плоских колебаний 25 см-1, для неплоских колебаний 17 см-1.
Опубликовано много неэмпирических расчетов силовых полей и колебательных спектров молекул небольшого и среднего размеров с использованием методики Пулея [145, 150 - 152]. Для малых молекул расхождения между вычисленными и экспериментальными частотами нормальных колебаний составили несколько обратных сантиметров [145, 150]. Для молекул среднего размера это расхождение несколько больше. Так, для 1Н-пиррол-2,5-диона среднеквадратичное отклонение вычисленных частот нормальных колебаний от экспериментальных величин составило 7 см-1 для плоских и 10 см-1 для неплоских мод [151], для имидазола эти величины равны соответственно 9 и 27 см-1 [152].
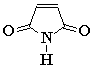
При подборе масштабных множителей или инкрементов естественно встает вопрос об их воспроизводимости в ряду разных молекул. В табл. 3.1 приведены корректирующие множители для 1Н-пиррол-2,5-диона.
Близкие значения коэффициентов из наборов 1 и 2 демонстрируют хорошую переносимость корректирующих множителей между молекулами, которые не являются родственными, но содержат однотипные валентные связи и углы. Может показаться, что вопрос о переносимости корректирующих множителей сходен с вопросом о переносимости определенных из опыта силовых постоянных, но в действительности происхождение ограничений универсальности в этих двух случаях совершенно различно; в первом случае в основе ограничения лежит существенная иррегулярность ошибок выбранного метода расчета, а во втором - реально существующая специфика силового поля у тех или иных исследуемых молекул, которая сама по себе чрезвычайно интересна и важна для химии и к тому же доступна для изучения квантовохимическими методами.
Таблица 3.1. Корректирующие множители для 1Н-пиррол-2,5-диона (неэмпирический расчет в базисе 4-21 ГФ)
|
Колебание |
Набор 1*1 |
Набор 2*2 |
Колебание |
Набор 1*1 |
Набор 2*2 |
|
Плоскиеколебания |
Плоскиеколебания |
||||
|
ν(N—H) ν(C—H) ν(C = O) ν(N—C) ν(C—C) ν(C = C) β(N—H) |
0,880 0,866 0,826 0,880 0,920 0,866 0,800 |
0,844 0,835 0,818 0,878 0,901 0,869 0,790 |
β(C—H) β(C = O) α(кольцо) |
0,800 0,836 0,800 |
0,785 0,875 0,793 |
|
Неплоские колебания |
|||||
|
γ(N—H) γ(C—H) γ(C = O) τ(кольцо) |
0,500 0,730 0,800 0,800 |
0,510 0,697 0,831 0,731 |
|||
*1 Усредненные величины по результатам расчетов для других (неродственных) молекул.
*2 Оптимальные величины для данной молекулы.
Большое разнообразие приемов и факторов корректирования является отражением субъективного подхода к этой проблеме. Наилучшее согласие с экспериментом дает, естественно, максимальная специализация поправок, хотя она неизбежно уменьшает, а в пределе сводит на нет значение самого первоначального расчета. Для экспериментатора наиболее ценной является информация о происхождении спектральных линий. По нашему мнению, такую информацию может дать лишь расчет со стандартными корректирующими множителями. Оптимизация корректирующих множителей, если она меняет первоначальное отнесение, вызывает сомнение. Эта процедура позволяет в 1,5 - 2 раза уменьшить среднее расхождение вычисленных и экспериментальных колебательных частот, но может внести дополнительные ошибки в их отнесение.
При использовании полуэмпирических методов следует учитывать, что в отдельных случаях они приводят к неверным знакам недиагональных коэффициентов, в частности, для недиагональных силовых постоянных, характеризующих взаимодействие связей С—Н и смежных валентных связей, однако абсолютная величина этих постоянных мала и поэтому они слабо сказываются на значениях частот, но от них зависит относительное расположение линий валентных колебаний в группах СН2 и СН3.
Для несмежных связей коэффициенты K(i,j) крайне малы, поэтому многие авторы при расчетах колебательных частот не принимают их во внимание. В некоторых работах высказывалось предположение, что в сопряженных системах типа
недиагональные коэффициенты, характеризующие взаимодействие кратных связей, аномально велики [132, 153, 154]. Однако результаты квантовохимических расчетов показали, что в сопряженных системах большую величину имеют коэффициенты, характеризующие взаимодействие смежных двойных и одинарных связей, причем расчет правильно воспроизводит большие значения величины расщепления полносимметричного и антисимметричного колебаний кратных связей, наблюдаемые экспериментально.
Напомним, что именно на основе этих экспериментальных данных было сделано неверное заключение в работах [132, 153, 154]. Их авторы использовали значения недиагональных силовых постоянных для смежных связей, заимствованные из несопряженных систем. При этом им пришлось взять аномально большие значения для недиагональных силовых постоянных, характеризующих взаимное влияние кратных связей, ибо в противном случае не удавалось воспроизвести экспериментально наблюдаемые величины расщепления полносимметричного и антисимметричного колебаний кратных связей. У авторов указанных работ не было каких-либо оснований для увеличения значений недиагональных коэффициентов взаимного влияния смежных одинарной и кратной связей. Это удалось сделать только с помощью квантовохимических расчетов [145, 155].
Квантовохимические расчеты колебательных частот широко используются для интерпретации экспериментальных данных и предсказания спектров соединений, которые не удается синтезировать или выделить из смеси других веществ или изомеров. Так, в ряде работ сделаны попытки изучить колебательные спектры изомеров, которые присутствуют в небольших концентрациях в равновесной системе. В цитированной выше работе [150] приведены интересные данные о спектрах S-цис-конформеров бутадиена и глиоксаля, относительно которых имелись лишь очень ограниченные и ненадежные сведения. По полученным данным, силовые постоянные валентных связей у S-цис- и S-транс-конформеров практически одинаковы, за исключением К(С—С), значение которой у цис-глиоксаля заметно меньше, чем у транс-конформера., за счет чего ωС=С ниже на 100 - 200 см-1. Коэффициенты взаимодействия во всех случаях малы, но в то же время у S-цис-формы в противоположность S-транс-форме частота симметричного колебания связей С-С должна быть ниже частоты антисимметричного. Детальная информация получена для поворотных изомеров муравьиной кислоты [156]. Спектр S-цис-конформера
был неизвестен, и расчет дал предсказания, имеющие значение для эксперимента. Применение неэмпирического метода с довольно большим базисом привело к следующим значениям силовых постоянных К(мдин/А) для S-цис-(S-транс-)конформера:
|
Связь |
S-цис |
S-транс |
|
С=О С—О О—Н С=О, С—О |
14,6 6,9 8,8 1,9 |
14,1 7,2 7,9 1,3 |
В соответствии с различиями в жесткости связей у S-цис-формы ωС=О должно быть выше на 50 см-1, а ωО-Н - на 200 см-1 по сравнению с соответствующими частотами S-транс-формы.
У некоторых аминокарбонильных соединений с сопряженными связями С=С и С=О в ИК-спектрах, кроме обычно наблюдаемых полос с частотами около 1650 см-1 (колебание связи С=О) и около 1570 см-1 (колебание связи С=С) появляется третья полоса в области около 1610см-1, происхождение которой не было ясно. Расчеты колебательных частот аминопропеналя [156] указывают на принадлежность этой полосы второму изомеру и вместе с тем на то, что К(С=О) увеличивается при транс-цис-изомеризации с 15,0 мдин/А (S-цис-изомер) до 16,1 мдин/А (S-транс-изомер), а К(С=С) при этом меняется мало.
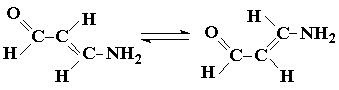
S-транс S-цис
Неэмпирический расчет силовых постоянных для метиламина и ряда других простых молекул [157—161] позволил дать отнесение некоторых линий, в отношении которых высказывались противоречивые мнения. Так, было установлено, что полоса 777 см-1 в колебательном спектре HNCO относится к неплоскому колебанию, а в молекуле гидроксиламина полоса крутильного колебания должна лежать в области около 1300 см-1.
Представляют интерес оценки жесткости связей атома металла у солей в отсутствие межмолекулярных взаимодействий. По данным расчета [162], коэффициент K(Li—С) в изолированной молекуле LiCN должен быть в 3 - 4 раза ниже, чем у связей с цианогруппой в молекулах HCN, ClCN и Н3ССN.
Согласно данным работы [134], силовые постоянные существенно различаются у связей С—Н, по-разному ориентированных по отношению к неподеленной паре электронов соседнего гетероатома. По расчету силовых постоянных и частот сделаны предсказания спектра винилборана, практически недоступного для эксперимента [163].
В связи со сказанным выше следует подчеркнуть, что результаты квантовохимических расчетов пока не могут охарактеризовать силовое поле молекулы и предсказать частоты нормальных колебаний с точностью эксперимента, однако они весьма полезны для отнесения линий, а совокупность данных расчета и эксперимента приводит к более полному и достоверному описанию спектра. При этом вычисленные относительные величины имеют большую достоверность и могут помочь при интерпретации сравнительно небольших различий в частотах сходных молекул, что не всегда удается сделать другими методами.