2.2. СОЛЬВАТОННАЯ МОДЕЛЬ
Эта модель была предложена Джано [85] и Клопманом [86]. В ее основе лежит предположение, что с каждым атомом растворенного соединения связан индуцированный в растворителе электрический заряд - сольватон. Заряд сольватона по абсолютной величине равен заряду на атоме, который его индуцировал, но имеет противоположный знак. Сольватоны не взаимодействуют между собой. Энергия взаимодействия между сольватоном С и атомом А вычисляется по следующей формуле:
EСА=qCqAе2(ε - 1)/(2rε)
где r — радиус атома А, если сольватон С индуцирован зарядом данного атома, или расстояние между атомами А и В, если сольватон индуцирован зарядом атома В;
qCи qА — заряды сольватона С и атома А в единицах заряда электрона;
е - заряд электрона; ε - диэлектрическая проницаемость среды.
Чаще для оценки ЕСА используют аналогичное выражение с кулоновскими интегралами, взятыми из квантовохимического расчета, т.е. е2/r полагается равным γАА, если сольватон С индуцирован зарядом на атоме А, или γАB, если сольватон С индуцирован зарядом атома В (γАAи γАB— одно- и двухцентровые кулоновские интегралы соответственно; см.разд. 1.2). Для энергии сольватации в этом приближении можно записать следующее выражение, где сумма берется по всем атомам растворенной молекулы:
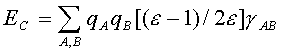
В качестве примера использования сольватонной модели для изучения механизмов органических реакций рассмотрим результаты работы [87], в которой изучена Z,E-изомеризация α,β-дизамещенных α,β-непредельных альдегидов.
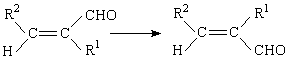
Расчеты были проведены методами ППДП/2 и МЧПДП/3. Эта реакция связана с внутренним вращением вокруг двойной связи С=С и в газовой фазе идет с очень высоким активационным барьером 200 - 250 кДж/моль. Преодолеть такой активационный барьер при нормальных условиях очень трудно, поэтому скорость газофазной реакции будет ничтожно мала.
Для реакции в растворе можно предложить следующие два механизма катализа, которые позволяют снизить активационный барьер.
1. Если растворитель обладает кислотными свойствами, то становится возможной протонизация исходного соединения, после чего по данным квантохимических расчетов активационный барьер реакции должен уменьшиться до 105 - 110 кДж/моль.
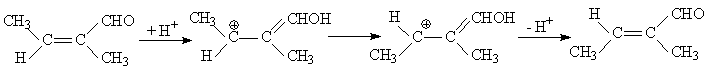
Учет влияния растворителя с использованием сольватонной модели (величина ε полагалась равной 5 и 10) приводит к дополнительному понижению высоты активационного барьера, и он становится равным 75 - 85 кДж/моль. Реакции с таким активационным барьером могут проходить при нормальных условиях. Таким образом, если растворитель обладает кислотными свойствами, то он может существенно ускорить реакцию Z,E-изомеризации α,β-дизамещенных α,β-непредельных альдегидов.
2. Если растворитель способен катализировать перенос атома Н от группы СН3 к СО, то становится возможным второй механизм увеличения скорости изомеризации:
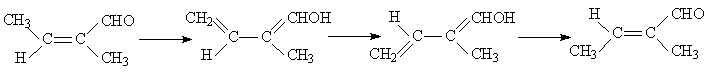
Высота активационного барьера изомеризации, проходящей по второму механизму, определяется высотой активационного барьера переноса протона от группы СН3 к карбонильному кислороду. Эта реакция, согласно результатам квантовохимических расчетов в газовой фазе, требует затраты около 65 кДж/моль. Учет сольватации повышает эту величину до 80 кДж/моль. Если растворитель обладает хорошей способностью к переносу протона (такой же, как вода), то высота активационного барьера будет близка к тепловому эффекту реакции, т.е. активационный барьер будет немногим выше 80 кДж/моль. С таким активационным барьером реакция может проходить при нормальных условиях.
Сольватонная модель проста и экономична. Однако она обладает рядом серьезных недостатков. Самый неприятный из них заключается в том, что сольватоны локализуются в непосредственной близости от атомов растворенной молекулы, которые их индуцировали, т.е. фактически проникают внутрь молекулы. В случае ионов сольватонная модель учитывает изменение энергии сольватации только за счет изменения степени делокализации заряда, хотя в действительности определяющим фактором в данном случае является их объем. Если изменение степени делокализации заряда коррелирует с изменением объема иона, то сольватонная модель будет давать правильные результаты. Из этого становятся ясными ограничения области ее применения. В частности, сольватонная модель не может учесть эффекты экранирования заряда неполярными группами или молекулами реагентов. По этой причине ее нельзя применять в комбинации с приближением "супермолекулы" при необходимости учесть эффект специфической сольватации.
В заключение настоящего раздела отметим еще одно важное обстоятельство. Выше было указано, что заряды на атомах, вычисленные разными методами, сильно различаются. Из-за этого сольватонная модель, будучи примененной в рамках разных методов, дает разные результаты. Она неплохо работает с методом ППДП/2. Мы пробовали использовать ее также в расчетах методами МЧПДП/2, МЧПДП/3 и МПДП, но результаты получились гораздо хуже.